
Sommaire
L’hépatite autoimmune (HAI) est une maladie inflammatoire du foie caractérisée par la présence d’autoanticorps (AAc) sériques, d’une hypergammaglobulinémie polyclonale et d’une infiltration lymphocytaire périportale qui n’est pas due à une autre cause (médicamenteuse, virale ou toxique).
Les HAI sont des maladies rares dont l’incidence annuelle est d’environ 1,9/100 000 habitants et la prévalence de 16,9 / 100 000. Elles représentent moins de 6 % des hépatites chroniques en France. La prévalence, comme pour celle d’autres maladies auto-immunes, varie selon un gradient géographique nord-sud. En effet, il existe une fréquence plus élevée en Europe du Nord que l’on peut expliquer par l’association avec l’haplotype HLA A1-B8-DR3.
La maladie peut débuter à tout âge, mais elle est particulièrement fréquente entre 10 et 30 ans et de 40 à 50 ans. Elle touche à la fois les hommes et les femmes, mais il existe une nette prédominance féminine selon un ratio de 4 pour 1.
Classification et auto-anticorps
La technique de l’immunofluorescence indirecte sur foie/rein/estomac de rat permet la détection de l’ensemble des auto-anticorps d’intérêt à l’exception des anticorps anti-soluble liver antigen (anti-SLA). Elle nécessite cependant l’utilisation de techniques de confirmation (ELISA, Dot-blot…) qui se sont considérablement développées grâce à l’identification des principales cibles antigéniques reconnues par ces anticorps. Actuellement, l’identification des différents auto-anticorps présents dans le sérum des patients permet de classer les HAI en deux types principaux : HAI de type 1 et de type 2.
Hépatite autoimmune de type 1
L’hépatite autoimmune de type 1 (HAI-1) est caractérisée par la présence d’anticorps anti-muscle lisse (anti-ML) de spécificité anti-câble d’actine, d’anticorps anti-nucléaires (ANA) et d’anticorps anti-SLA. 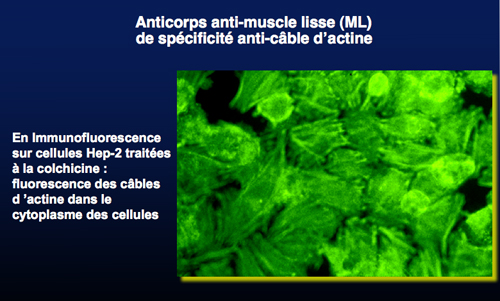
 Les anticorps anti-actine sont présents dans plus de 85% des HAI-1 avec une spécificité de 80%. Ils sont en effet présents dans d’autres pathologies : hépatites virales, médicamenteuses, pathologies autoimmunes non hépatiques (connectivites, maladies endocriniennes). Le typage de l’anticorps anti-muscle lisse par immunofluorescence indirecte sur cellules Hep-2 traitées à la colchicine, dot-blot ou ELISA est obligatoire, un anticorps anti-muscle lisse non actine n’ayant aucune spécificité vis à vis des HAI-1.
Les anticorps anti-actine sont présents dans plus de 85% des HAI-1 avec une spécificité de 80%. Ils sont en effet présents dans d’autres pathologies : hépatites virales, médicamenteuses, pathologies autoimmunes non hépatiques (connectivites, maladies endocriniennes). Le typage de l’anticorps anti-muscle lisse par immunofluorescence indirecte sur cellules Hep-2 traitées à la colchicine, dot-blot ou ELISA est obligatoire, un anticorps anti-muscle lisse non actine n’ayant aucune spécificité vis à vis des HAI-1.
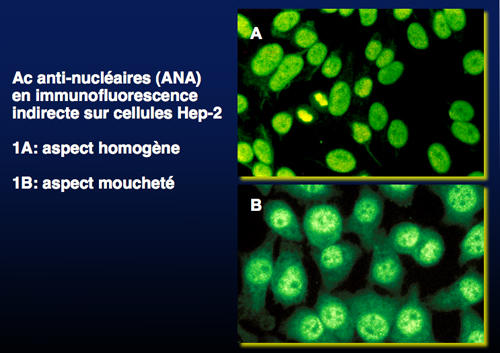
Les anticorps anti-nucléaires sont détectés dans 70% des HAI-1, responsables approximativement dans 35% d’une fluorescence homogène du noyau et dans 35% d’une fluorescence mouchetée. Ils sont présents de façon isolée dans 15% des HAI-1. Leurs cibles antigéniques sont multiples. Notre groupe a identifié récemment une ribonucléoprotéine, l’isoforme hnRNPA2/B1, qui intervient dans le transfert de l’ARNm du noyau vers le cytosol ainsi que dans le maintient de sa structure tridimensionnelle. D’autres cibles telles que la chromatine, les protéines histones, les lamines, des protéines nucléaires solubles sont aussi décrites dans cette pathologie hépatique.
Enfin les anticorps anti-SLA sont présent dans 15 à 30% des HAI-1, 20% des hépatites cryptogéniques qu’ils permettent de reclasser en HAI-1 et 20% des formes mixtes. Leur spécificité est excellente (supérieure à 99%). Une seule équipe a décrit la présence d’anticorps anti-SLA dans les HAI-2 et les cholangites sclérosantes primitives mais en utilisant comme antigène la protéine recombinante tRNP(ser)sec avec une technique radioimmunologique. Ces anticorps pourraient être de mauvais pronostic et ont été décrits dans les HAI de novo et les récidives d’HAI-1 après transplantation hépatique.
L'hépatite autoimmune de type 2
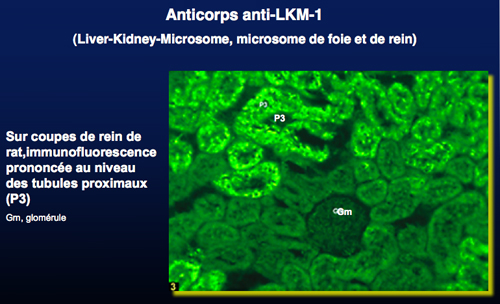
L’HAI de type 2 (HAI-2) est caractérisée par la présence d’anticorps anti-LKM1 (Liver-Kidney-Microsome) ou anti-CYP2D6.
Ils sont présents dans 85% des HAI-2, mais se rencontrent aussi dans 5% des hépatites virales C où ils reconnaîtraient d’autres épitopes. La fréquence des anticorps anti-liver cytosol 1 (anti-LC1) ou anti-formiminotransferase cyclodésaminase varie de 30 à 50% lorsque l’antigène utilisé est un extrait de cytosol hépatique, et peut aller jusqu’à 70% lorsqu’on utilise la protéine recombinante. Dans 10% des cas, les anticorps anti-LC1 représentent le seul marqueur immunologique des HAI-2. Une étude récente a montré que les patients ayant une HAI-2 avec anticorps anti-LC1 isolés présentent moins de maladies autoimmunes associées. Leur spécificité n’est pas parfaite, il peuvent être détectés chez 0.5% des patients atteints d’infection virale C. Contrairement aux marqueurs de l’HAI-1, l’intérêt pronostic est certain: en effet, les titres des anticorps anti-LKM1 et anti-LC1 varient avec le stade de la maladie et le traitement utilisé.
D’autres auto-anticorps ont également été identifiés dans les HAI et en particulier des auto-anticorps dirigés contre le cytoplasme des polynucléaires neutrophiles (ANCA), responsables d’un aspect dit p-ANCA atypique. Le terme d’ANCA est en fait impropre car leurs cibles moléculaires, mal connues, sont d’origine nucléaire, expliquant le terme NANA (nuclear associated neutrophil antibodies) quelquefois employé. Une protéine de 50 kD de la membrane nucléaire a récemment été proposée. 40 à 50% des patients avec une HAI de type 1 sont positifs pour les p-ANCA atypiques tandis que la plupart des patients présentant une HAI-2 sont négatifs pour ce type d’anticorps. Cependant, ces anticorps sont présents dans 50 à 80% des cholangites sclérosantes primitives (CSP) et dans les maladies inflammatoires de l’intestin sans CSP.
Dans 80% des HAI-1, des auto-anticorps anti-récepteurs à l’asialoglycoprotéine sont également présents, mais on les trouve aussi dans d’autres pathologie hépatiques en particulier virales et alcooliques, ils ne sont aucunement spécifiques de l’HAI. Ils en représenteraient cependant un marqueur de gravité.
Enfin, les anticorps anti-mitochondries de type 2, dirigés contre les complexes 2 oxo-déshydrogénasiques présents dans plus de 95% des cirrhoses biliaires primitives chez l’adulte, peuvent être détectés de façon exceptionnelle chez l’enfant où ils apparaissent alors comme des marqueurs d’HAI.
Signes cliniques, biologiques et évolution
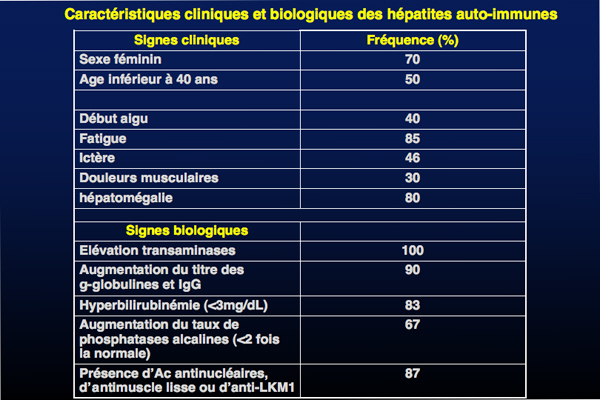
Les HAI touchent essentiellement la femme (70% des cas) et 50% des patientes affectées ont moins de 40 ans. La révélation de la maladie se fait habituellement entre 30 et 50 ans.
Bien que les antécédents familiaux soient rares, la maladie a déjà été rapportée chez des jumeaux et les parents de premier et deuxième degré. De plus les parents de premier degré ont volontiers un taux d’immunoglobulines anormale (47% des cas), des auto-anticorps (42%) et une hypergammaglobulinémie (34%).
La maladie est diagnostiquée grâce à un ensemble de signes cliniques non spécifiques, tels que l'asthénie (85% des cas), un ictère d’intensité variable (80%), une hépatomégalie (80%) ou des hépatalgies (50%). Dans environ 30% des cas, le mode de présentation est aigu et peut mimer un tableau d’hépatite virale ; dans les autres cas, le début est plus insidieux et la maladie n’est pas reconnue avant un stade très évolué. Dans la moitié des cas, des manifestations dysimmunitaires extra-hépatiques telles que des arthralgies, une dysthyroïdie ou un diabète insulinodépendant peuvent être associés. Plus rarement et en particulier dans les formes d’HAI-2 de l’enfant, la maladie peut se déclarer sous une forme subfulminante ou fulminante.
Dans le bilan biologique, une cytolyse hépatique avec des taux de transaminases élevés (entre 5 et 10 fois supérieurs à la valeur normale) et une hypergammaglobulinémie polyclonale (2 à 3 g/dL) à prédominance d’IgG sont classiquement observées. Une élévation du taux de phosphatases alcalines est détectée chez 80% des patients : de l’ordre de 2 fois la valeur normale chez 33% des patients et plus de 4 fois la normale dans 10% des cas ; dans ces cas la présence d’une forme frontière est suspectée.
L’évolution se fait par poussées successives, parfois spontanément résolutives, laissant comme séquelle une fibrose hépatique pouvant évoluer vers une cirrhose. Celle-ci peut se constituer de façon insidieuse avec un diagnostic tardif porté à ce stade dans 25% des cas; 40 % des patients sont au stade de cirrhose après 10 ans de suivi. Le risque d’apparition d’un carcinome hépatocellulaire existe mais est très faible.
Signes histologiques
L’HAI est caractérisée par une inflammation lympho-plasmocytaire portale et péri portale avec nécrose d’intensité variable et avec la présence ou non d’une hépatite lobulaire. La présence d’une atteinte centrolobulaire sans atteinte portale a aussi été rapportée.
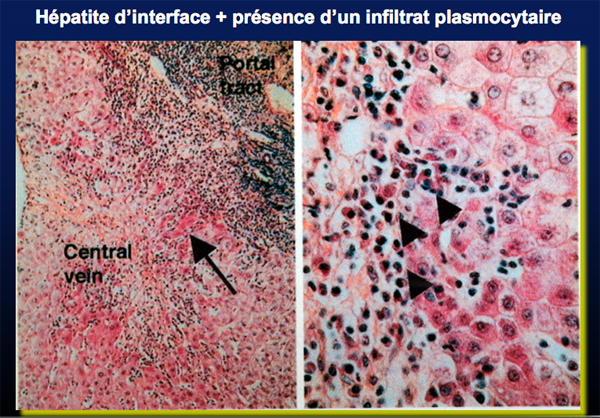
Les formes frontières
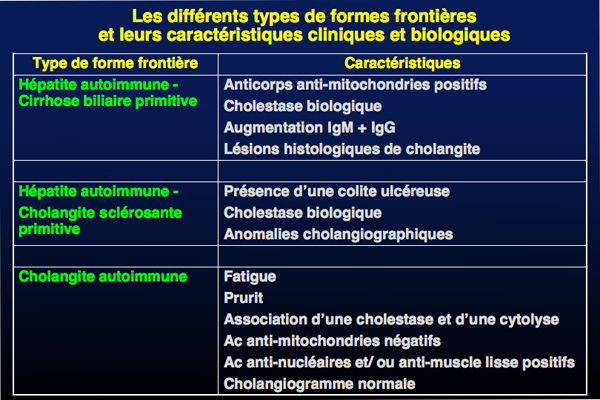
Les formes frontières ou "syndrome de chevauchement" associent à des degrés divers des aspects de cirrhose biliaire primitive, de cholangite sclérosante primitive et d’HAI.
La présence conjointe d’une séropositivité pour les anticorps anti-mitochondries et d’un syndrome de cholestase clinique, biologique et histologique chez une patiente atteinte d’HAI suggère l’existence d’une forme frontière hépatite autoimmune - cirrhose biliaire primitive. Cette forme concerne 5% des patients atteints d'hépatite autoimmune et 19% des patients avec de cirrhose biliaire primitive.
Pour les formes où l'hépatite autoimmune est prédominante, le traitement repose sur la corticothérapie, tandis que pour les formes où la cirrhose biliaire primitive est prédominante, il repose sur l’acide ursodesoxycholique.
Les patients qui présentent la forme frontière hépatite autoimmune - cholangite sclérosante primitive ont des signes biologiques et histologiques d’HAI avec des signes histologiques et radiologiques de cholangite sclérosante primitive. Le traitement est empirique et dépend de la forme prédominante : une corticothérapie doit être tentée pour les formes hépatiques prédominantes et l’acide ursodexoxycholique pour les formes cholestatiques bien que ce traitement n’ait pas juqu’à présent fait preuve de son efficacité dans les cholangites sclérosantes primitives.
Traitement
Le but du traitement est de limiter l’activité de la maladie et les rechutes ultérieures de manière à diminuer le risque d’évolution vers la cirrhose et ses complications.
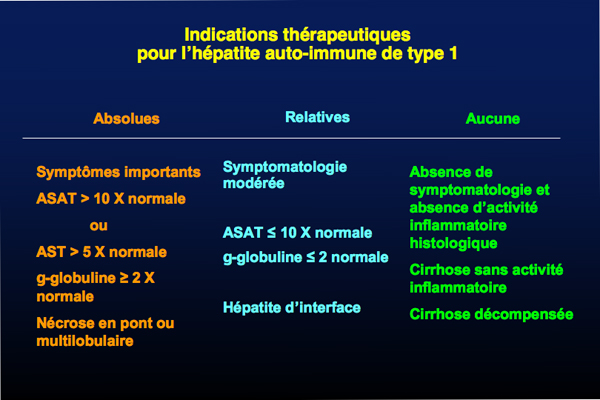
Un traitement est nécessaire en cas :
-
de forme fulminante, qui est une urgence thérapeutique ;
-
d’hépatite subaigüe avec un taux de transaminases entre 5 et 10 fois la normale et des gammaglobulines supérieures à 2 fois la normale ;
-
des signes cliniques invalidants ;
-
d'une atteinte histologique modérée à sévère (définie par le degré de l’atteinte inflammatoire et de la nécrose).
Une activité histologique minime peut justifier l'absence de traitement car celui-ci, dans cette indication, n’a pas prouvé son efficacité sur la survie à long terme.
En cas de non traitement, le taux de mortalité peut dépasser 80%, en particulier chez les patients avec un taux de transaminase supérieur à 5 fois la valeur normale et un taux d'IgG supérieur à 2 fois la normale. Les facteurs de mauvais pronostic sont : le jeune âge, la positivité des anticorps anti-LKM1, l’allongement de l’INR, un taux élevé de bilirubine et une activité histologique importante.
Les modalités thérapeutiques

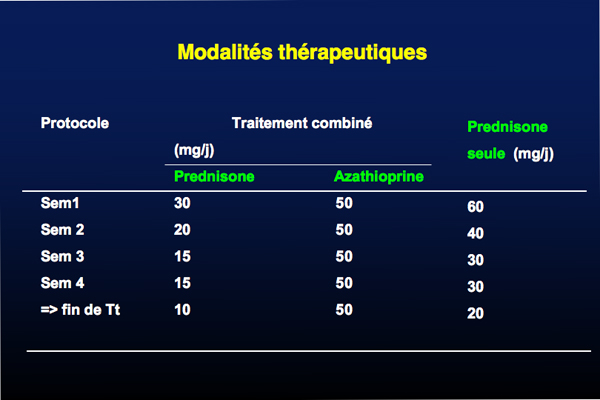
La corticothérapie seule ou en association avec l’azathioprine est efficace dans le traitement des HAI. Plus de 80% des patients suivant ce traitement immunosuppresseur ont une réponse complète, avec toutefois la présence d’effets secondaires qui peuvent être très importants. La combinaison prednisone–azathioprine est préférée car elle diminue la fréquence des effets secondaires de la corticothérapie.
Traitement Initial
La corticothérapie associée à l’azathioprine (prednisone: 1mg/kg/jour maximum 60 -mg/jour; azathioprine: 50 à 100 mg/jour) constitue le traitement de choix. Le but de cette bithérapie est de diminuer rapidement les doses de corticoïdes une fois la normalisation des transaminases obtenue. La baisse des doses s’effectue par paliers de 5 mg toutes les 2 semaines lorsque le taux de transaminases est inférieur à 2 fois la normale.
Les candidats à une bithérapie sont des femmes ménopausées, des patientes avec une ostéoporose ou une ostéomalacie, ou présentant une obésité, un diabète ou une hypertension artérielle. Les candidats à une monothérapie par corticoïdes sont les patients qui présentent une cytopénie et les patientes qui désirent ou qui sont en cours de grossesse.
Traitement d’entretien
La baisse des doses de corticoïdes s’effectue par paliers successifs, de façon à donner la dose minimale pour maintenir la normalisation des taux de transaminases, cette dose s'établit généralement entre 5 et 15 mg de corticoïdes par jour. Ce traitement doit être poursuivi pendant plusieurs années (5 à 10 ans en moyenne) jusqu’à ce que l’interruption des corticoïdes n’entraîne pas de rechute. Celle-ci se produit dans environ 50% des cas. C’est pourquoi, chez ces malades, le traitement corticoïde doit être maintenu longuement afin de prévenir une rechute.
Dans le cas où l’interruption du traitement par corticoïdes est possible, il est préférable de maintenir l’azathioprine seule pendant 1 ou 2 ans car ceci permet une interruption avec un taux de rechute moindre.
Une biopsie de contrôle doit être réalisée à la fin du traitement dans le but d’apprécier l’amélioration histologique. Sans amélioration, la poursuite du traitement immunosuppresseur doit être discutée.
En cas de grossesse, lorsque la bithérapie est déjà instituée, il est conseillé de ne pas arrêter l’azathioprine sous réserve d’une surveillance très rigoureuse.
En cas de non réponse
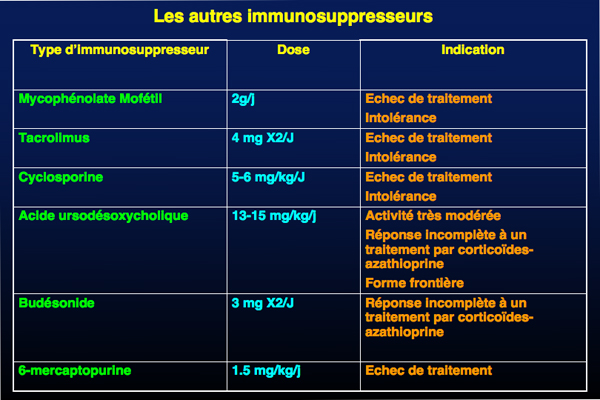 20% des malades ne répondent pas au traitement et l’absence de bénéfice thérapeutique s’observe habituellement dans les 3 premiers mois. D’autres alternatives thérapeutiques doivent être proposées après l’essai d’une augmentation des doses de corticoïdes et d’azathioprine.
20% des malades ne répondent pas au traitement et l’absence de bénéfice thérapeutique s’observe habituellement dans les 3 premiers mois. D’autres alternatives thérapeutiques doivent être proposées après l’essai d’une augmentation des doses de corticoïdes et d’azathioprine.
D’autres immunosuppresseurs tels que 6-mercaptopurine, cyclosporine, tacrolimus et mycépholate mofétil (MMF) peuvent être utilisés en deuxième intention. Le MMF paraît très efficace en terme de réponses biologique et histologique chez des patients en rechute après l’association corticoïdes-azathioprine.
L’acide ursodesoxycholique associée à la corticothérapie, apporte un bénéfice au moins biologique à des doses de 750 mg/j. Ce traitement est conseillé dans les formes frontières.
La transplantation hépatique
![]()
La transplantation hépatique est actuellement indiquée en cas d’hépatite (sub) fulminante résistant à un traitement à fortes doses par corticoïdes ou en cas de cirrhose accompagnée de complications sévères. L’hépatite auto-immune représente environ 5% des indications de transplantation pour cirrhose. Le taux de survie après transplantation est de 80% en moyenne à 2 ans. La maladie peut récidiver jusque dans 40% des cas ; cette récidive, qui peut se manifester sous différentes formes (parfois sévères), serait favorisée par une immunosuppression insuffisante ou par un greffon présentant l’haplotype HLA DR3
Conclusion
Les hépatites auto-immunes représentent un exemple de maladie auto-immune pour laquelle, bien qu’il existe des critères diagostiques précis et une thérapeutique immunosuppressive efficace, de nombreuses interrogations subsistent telle que : la nature des auto-antigènes, la nature des mécanismes effecteurs, la compréhension des formes frontières, la prise en charge des formes corticosurrénales.
Une meilleure compréhension des mécanismes immunologiques impliqués dans cette pathologie permettra d’obtenir des traitements mieux ciblés et plus efficaces.